|
首页
>
资讯
>
国内药政每周导读:2022医保国谈启动,CDE再推儿童药开发,重庆局发变更问答,59批参比目录公示
出自识林
国内药政每周导读:2022医保国谈启动,CDE再推儿童药开发,重庆局发变更问答,59批参比目录公示
2022-06-20
【政策与监管综合】
6.13,医保局征求《2022年国家基本医疗保险工伤保险和生育保险药品目录调整工作方案》及相关文件意见
去年的“灵魂砍价”和“天价救命药(诺西那生)大幅降价进医保”等等新闻仿佛还历历在目,又到2022年医保目录调整之时。
简单回顾2021年的成果。2021年11月9日-11日,为期3天的2021年国家医保目录药品谈判在北京进行。此次医保谈判,共计74种药品新增进入目录,11种药品被调出目录。从谈判结果看,最终94种药品(目录外67种,目录内27种)谈判成功,总体成功率80.34%。目录外67种药品平均降价61.71%。2021年,国家医保目录内药品数量再创新高,达2860种,其中西药1486种,将于2022年1月1日执行。
同去年对比,医保谈判流程与关键政策基本一致,几个值得关注的要点:
- 利好儿童药物和罕见病药物放宽条件,申报条件中包括今年6月30号之前获批的儿童药品和罕见病药品,但并不要求在2017年之后获批。
- 时间安排是:5-6月准备,7-8月申报,8月专家评审,9-10月谈判,11月公布结果。若按照此时间线,会比去年稍微早一些。
- 附件《谈判药品续约规则(征求意见稿)》明确续约规则,利好医保稳定品种,长期来看利好所有品种在达到稳定状态后,维持稳定的医保价格。
- 续约的三个规则是:
- 规则一,纳入常规目录管理:非独家产品及2018年谈判进入目录且连续两个协议周期均未调整支付标准和支付范围的独家药品可以纳入常规目录管理,不再进行定期续约谈判;
- 规则二,简易续约:对于独家产品、且协议期医保基金实际支出未超过预估值200%、且预计未来的预算增幅合理的产品,进行简易续约,根据实际支出超过预估值的比例进行梯度价格下调;
- 规则三,重新谈判:对于不符合纳入常规目录及简易续约的独家产品,进行重新谈判。
符合申报条件的通用名药品获批企业均可提交报价,并与医保测算专家提出的医保支付意愿价格比较,低于医保支付意愿价格的最低价即为该通用名药品的最终医保支付价,若企业报价均高于医保支付意愿价格,则不纳入医保。
根据公号“风云药谈”整理,目前符合医保文件的药品接近170个,其中2017年1月1日以后获批符合相应条件产品有62个,2021年申报未进入医保目录产品达100个。
2021年医保谈判调整的范围要求是在2021年6月30前获批上市的新药,此后近一年来国内也有多款新药上市,值得关注的重磅产品如:
- PD-1:正大天晴/康方生物的派安普利单抗、誉衡生物/药明生物的赛帕利单抗、复宏汉霖的斯鲁利单抗;
- PD-L1:康宁杰瑞/先声药业/思路迪的恩沃利单抗、基石药业的舒格利单抗;
- CAR-T产品:药明巨诺靶向CD19的CAR-T产品瑞基奥仑赛注射液;
- ALK-TKI:武田新一代ALK酪氨酸激酶抑制剂(TKI)布格替尼(Brigatinib);
- JAK1抑制剂——艾伯维的乌帕替尼(upadacitinib)、辉瑞的阿布昔替尼;
- 罗氏的托珠单抗注射液(皮下注射)。
此外,还有一批去年未能谈判成功的药物,例如默沙东的帕博利珠单抗(K药)和BMS的纳武利尤单抗(O药),以及罗氏的阿替利珠单抗和阿斯利康的度伐利尤单抗;辉瑞的哌柏西利(商品名:爱博新®)、复星凯特的阿基仑赛注射液、罗氏的脊髓性肌萎缩症(SMA)罕见病药物利司扑兰、武田的维布妥昔单抗、信立泰的注射用重组特立帕肽等。
6.14,NMPA征求《关于鼓励企业和社会第三方参与中药标准制定修订工作有关事项的公告》意见
《公告(征求意见稿)》对企业和社会第三方参与中药标准制定修订工作的标准适用范围、适用对象、参与方式、工作要求、鼓励措施、保障措施等进行了明确。
本《公告》所称的中药标准是指国务院药品监督管理部门颁布的中药标准(包括中药材标准、中药饮片标准及中成药标准),以及各省、自治区、直辖市人民政府药品监督管理部门颁布的中药材标准和中药饮片炮制规范。
参与中药标准制修订的对象,药品上市许可持有人、中药生产企业、企业与科研机构组成的联合体、社会团体、独立的社会第三方机构均可参与中药标准工作。
【创新研发】
6.17,CDE征求《儿童用药口感设计与评价的技术指导原则(公开征求意见稿)》意见
口服给药是目前临床最常用的给药方式,口感是影响口服制剂临床应用的因素之一。儿童因其生理和心理发育特点,在不良感觉的耐受性方面有别于成人,口感不佳所导致的不良用药行为风险也相应增高,因此,儿童用药口感评价具有更强的临床意义与价值。
CDE继续不遗余力推进儿童药监管科学发展,除了本文,CDE还在同日发布《关于启动儿童用药技术审评临床专家咨询委员会遴选的通知》,向全国招募儿童用药专家参与临床技术审评。
识林企业用户可阅读主题词【儿童用药】,系列了解国内外相关法规指南。
【CMC与仿制药】
6.16,药典会征求《药包材检验规则指导原则》意见
继上周6月10日征求《药包材生物学评价与试验选择指导原则》意见之后,药典会再发本文,继续完善药包材的质量监管体系。
本文件给出了药包材生产企业、使用企业或其他相关第三方制定药包材检验规则的指导原则。监管部门在制定监督检验方案时可参考本文件的相关内容。因此,药企也应关注。
本指导原则从两个方面对药包材的检验规则进行了总体设计:
- 一是给出全生命周期的不同阶段应实施的检验类型,以及适用的检验项目。本文件将药包材的检验分为批检验、型式检验、对声称质量水平的评定检验。
- 二是对药包材的质量特性进行分级,推荐相应级别的质量水平限,并指导设计抽样检验方案。对于特定的质量特性,应从使用风险的角度来合理确定质量水平,并结合经济性确定检查水平。在保证科学性的前提下,使标准更易操作。
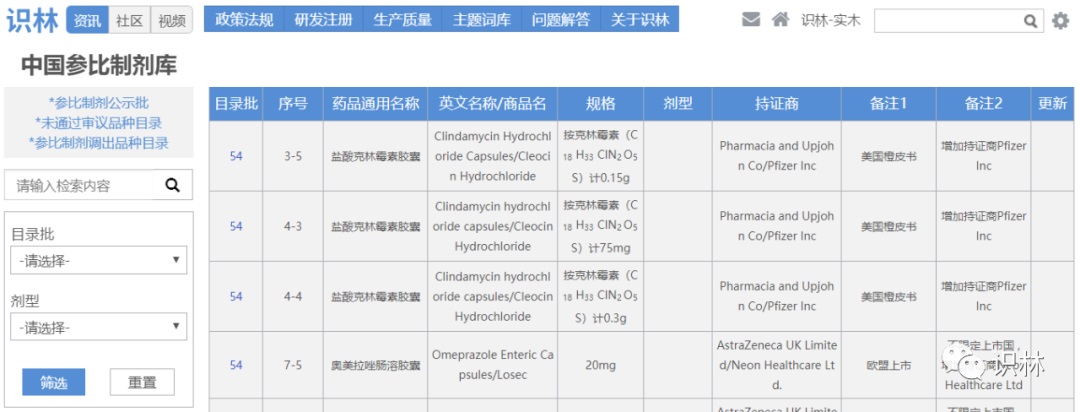
6.17,CDE征求《化学仿制药参比制剂目录(第五十九批)》(征求意见稿)意见
截至目前,参比制剂目录已公示至第五十九批,已发布至第五十四批。
识林“中国参比制剂库”可查阅全部内容。
【注册,审评,审批】
6.17,CDE药品审评报告和说明书更新,其中包括下列国产新药
注:仅列出国产创新药和改良型新药,供参考。企业用户可至识林“CDE药品审评报告”按发布时间查阅最新的上市药品信息。
【上市后监管】
6.15,辽宁省局发布《关于做好药品年度报告填报工作的通告》
NMPA于4月12日正式发布《药品年度报告管理规定》,形成了中国药品监管的闭环。
辽宁省局发布本文,提醒省内企业在8月31日前,按时提交首次年报。
省局要求与NMPA基本一致,有一处值得注意的是,《药品年度报告管理规定》的第十七条规定,“中药配方颗粒、疫苗等另有规定的,从其规定。”
而辽宁局明确提出“疫苗上市许可持有人在报送质量年度报告的同时,一并报送年度生产计划;原料药暂不需要报送年度报告;其他类型产品年度报告国家另有规定的,从其规定。”
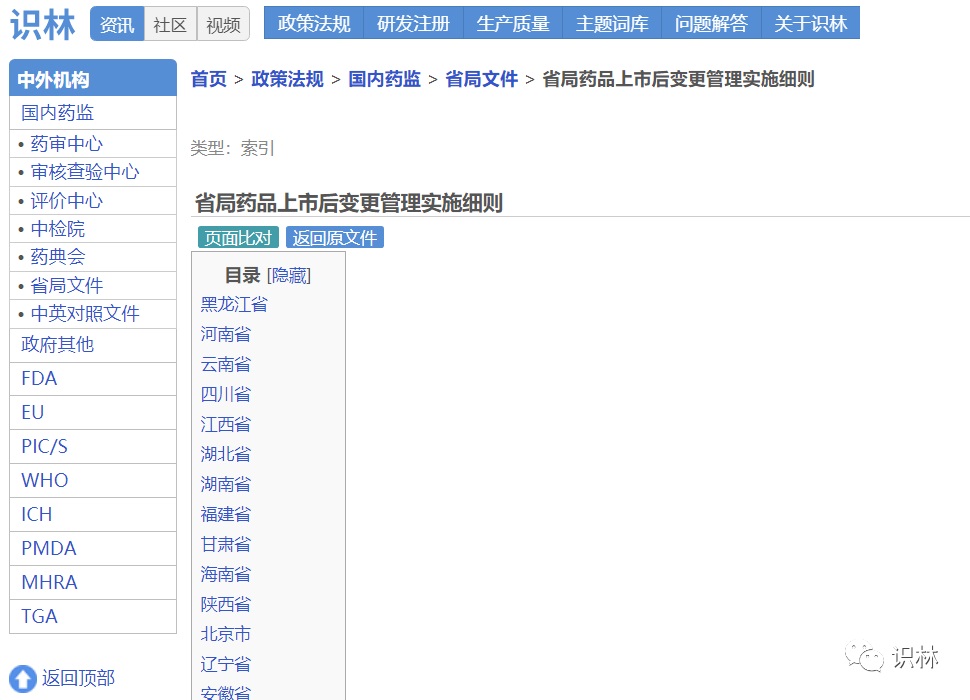
6.16,重庆局发布《药品上市后变更常见问题解答(二)》
本文包括12个问答,值得关注的是未在《已上市化学药品药学变更研究技术指导原则(试行)》中明确的内容,诸如:
- 二、某化药注射剂,删除或增加工艺中使用的活性炭,是否属于中等变更?
- 答:活性炭在注射剂中主要起吸附热原、脱色、助滤等作用,不论删除或增加对药品质量影响可能较大,一般归属制剂生产工艺的重大变更。
- 六、中等变更一般需提供不少于3个月的稳定性数据,无菌、微生物限度等指标不是每个时间点都进行考察,是否可以仅提供0月数据,不提供3个月的数据?
- 答:无菌、微生物限度等指标可以选择有代表性的时间点进行考察,应提供稳定性考察方案,明确无菌、微生物限度等指标的考察时间点,并承诺稳定性考察按照方案继续进行,直至确定的有效期,并在年报中报告。
- 十一、原研品说明书对适应症、用法用量等进行了调整,能否通过省局备案对自制品的说明书进行修订?
- 答:不能。该事项属于重大变更。
识林企业用户可查阅“省局药品上市后变更管理实施细则”页面,查找各省市局发布的变更指南和问答。
【新药批准和报产】
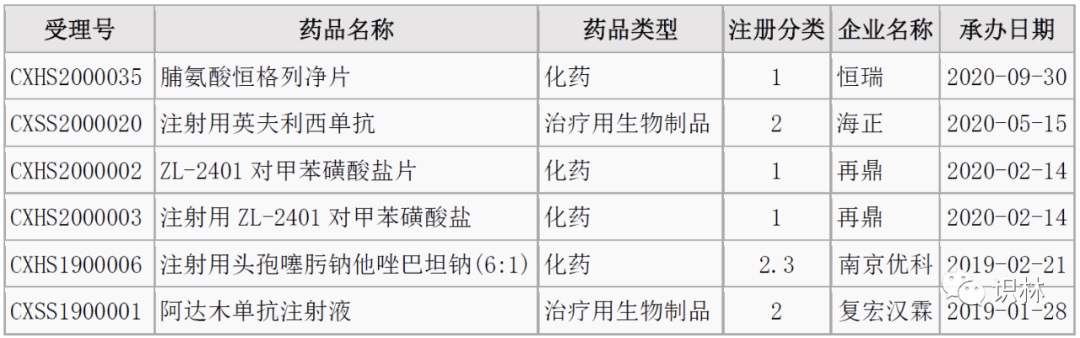
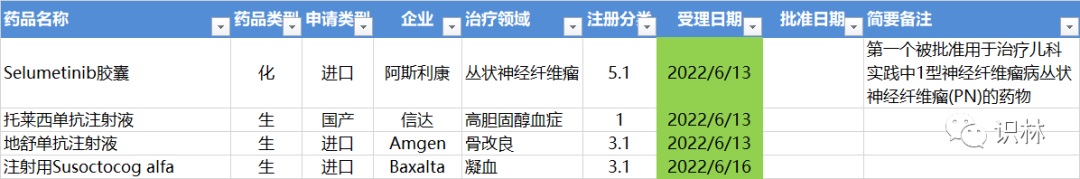
6.13-6.19,NMPA未发布新药批准,CDE受理4个新药上市
注:仅列出新药(包括改良型新药和生物类似药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
作者:识林-实木
识林®版权所有,未经许可不得转载
适用岗位及工作建议: - QA(质量保证):必读。需确保变更研究符合指导原则,监督变更实施过程,保证产品质量。
- 注册部门:必读。负责变更注册申请,确保申报材料符合要求。
- 生产部门:必读。根据变更指导原则调整生产工艺,确保生产过程合规。
- 研发部门:必读。参与变更研究工作,提供技术支持。
适用范围:
本文适用于已上市化学药品的药学变更研究,包括化学原料药和化学制剂。适用于不同注册分类的药品,包括创新药和仿制药。由中国药品监督管理局(NMPA)发布,适用于各类企业,包括Biotech、大型药企、跨国药企、CRO和CDMO等。 要点总结: - 变更研究主体责任:持有人/登记企业负责设计并开展变更研究,全面评估变更对药品的影响。
- 变更分类:根据对药品安全性、有效性和质量可控性的影响风险,变更分为重大变更、中等变更、微小变更。
- 关联变更:一项变更可能伴随或引发其他变更,需综合考虑各项变更研究工作的要求。
- 稳定性研究:变更后需进行稳定性研究,必要时增加研究批次或延长研究时间。
- 包装材料和容器变更:变更可能影响药品的保护性、功能性、安全性和质量,需进行相应的研究验证。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - QA:确保年度报告的真实性、准确性,并符合法规要求。
- 注册:了解年度报告的提交要求和流程,确保按时提交。
- 研发:参与年度报告中上市后研究及变更管理部分的撰写。
- 临床:提供上市后风险管理计划和相关数据支持。
文件适用范围:
本文适用于中国境内的持有人,包括创新药、仿制药、生物类似药、原料药等药品类型,由国家药品监督管理局发布,适用于大型药企、Biotech、CRO和CDMO等企业类别。 文件要点总结: - 年度报告责任主体:持有人负责年度报告的真实性和准确性,境外企业需指定境内代理人。
- 年度报告制度:持有人需建立并实施年度报告制度,包括填报、管理和审批流程。
- 信息收集与报告:年度报告应包含生产销售、上市后研究、风险管理等信息,并于每年4月30日前提交。
- 监督检查与整改:药品监督管理部门将对年度报告制度进行检查,不符合规定的需在规定时间内整改。
- 信息保密:未经持有人同意,不得披露商业秘密或未披露信息,除非涉及国家安全或重大公共利益。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |