|
首页
>
资讯
>
FDA 仿制药办公室发布 2019 年报
出自识林
2020-02-21
美国 FDA 仿制药办公室(OGD)于 2 月 19 日发布 2019 年度报告,这是其发布的第五份年报,报告在形式上与以往相比有较大变化,更加面向公众,以通俗的语言解释了 OGD 所做的事情,而不是简单的数据和资料的罗列。从指南篇幅上看,本次的报告更加注重研究方面的内容,对于 OGD 资助的研究及取得的成就方面有着更多介绍,关注研发的仿制药企业可着重看看这一部分及相关参考资料,想必会有所助益。
报告内容上一如既往地总结了 OGD 在过去一年中所取得的成绩,提供了有关批准和暂时批准的数据,仿制药使用者付费修正案(GDUFA II)的完成情况,以及其它监管行动,包括制定和发布的各种操作性文件、指南和政策。
下面我们来看看这份报告中的重点内容。
一些数据
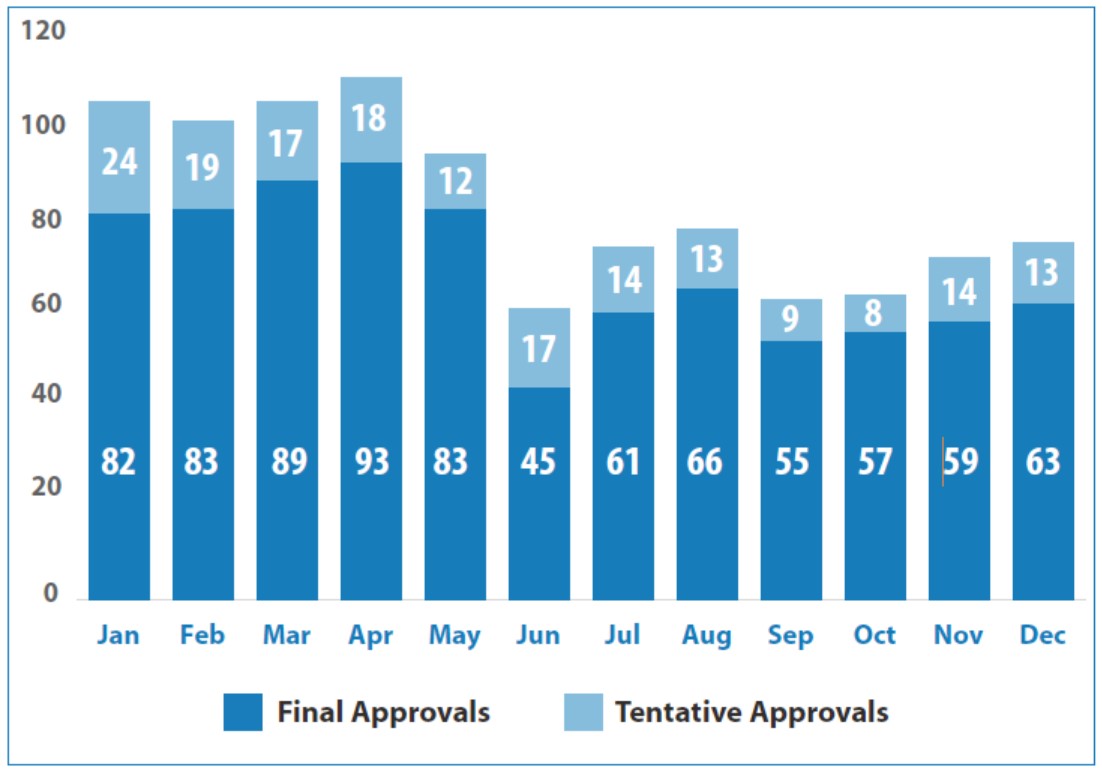
批准或暂时批准了 1014 件简化新药申请(ANDA)。其中 10%(107 件)为首仿药,11% (110 件)为复杂仿制药。发布了 2311 封完全回应函。回复了 3274 封受控函,再创历史新高。发布了 269 份具体产品生物等效性指南。
2019 年仿制药批准和暂时批准
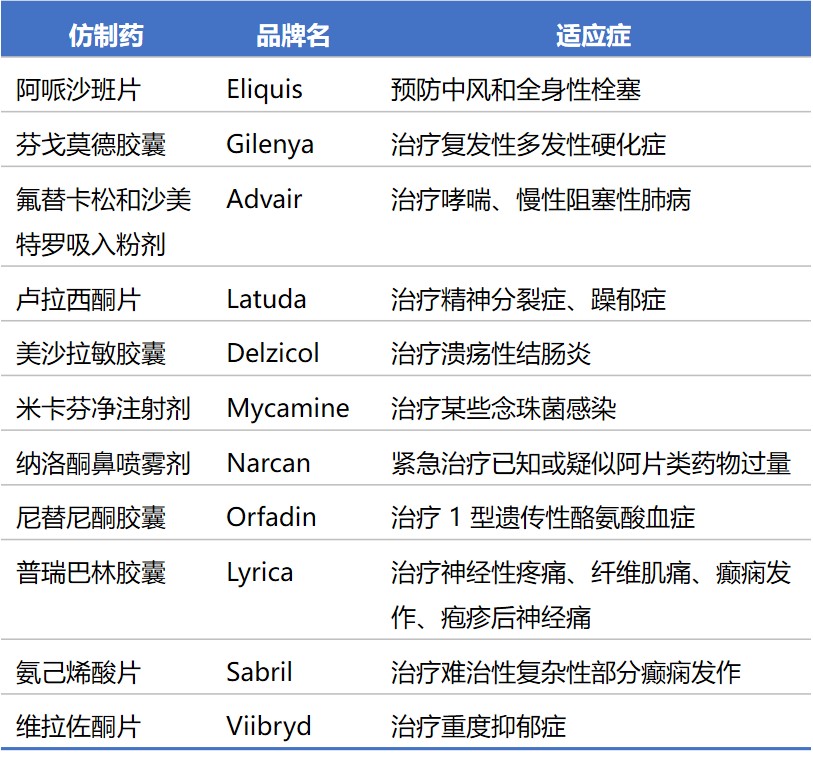
2019 年批准的重要首仿药
一些成绩
OGD 在提高首轮批准率和增加仿制药可及性的目标方面取得重大进展。OGD 继续满足或超越了其在 GDUFA 承诺函中的几乎所有时限。
2019 年,OGD 全球事务团队成功领导了关于“速释固体口服制剂生物等效性(BE)”的仿制药提案工作,并获得 ICH 管理委员会认可。ICH 将着手开发首个关注仿制药的 ICH M13 指南。
2019年,OGD 的临床安全监督人员(Clinical Safety Surveillance Staff,CSSS)在 OGD 内部和整个 CDER 展开工作,解决了与多个仿制非处方药和处方药中亚硝胺杂质有关的安全问题。还支持更改洛哌丁胺片的包装以促进安全使用。
药品竞争行动计划(DCAP)
2019 年,OGD 在 DCAP 的三个主要方面取得了重大进展:
1. 为提高仿制药开发、审评和批准流程的效率,发布以下指南,并更新 FDA 关于专利声明和适用性请愿的网页。
2. 在复杂仿制药方面最大限度地提高科学和法规清晰度
3. 封闭那些可以让品牌药公司钻空子延迟仿制药竞争的漏洞
重大研究成就
2019 年,仿制药计划在监管科学研究项目方面提供了约 2 千万美元的资金。授予了 14 项新合同和 3 项新资金,另外还有 2 项正在进行中的资金和 23 项合同开展监管科学研究。OGD 在 2019 年持续开展了 57 项外部研究合作,其中许多延续自往年的项目。
2019 年的重要研究成就包括:
- 发表地塞米松眼用混悬剂眼吸收模型的结果。
- 使用超高效液相色谱-质谱仪(UHPLC-MS)鉴定并定量了特立帕肽药物原料药和制剂的肽杂质谱。
- 与合作伙伴一道成功开发并验证了使用一系列内部合成的分支-PLGA(聚乳酸-乙醇酸共聚物)标准的分析技术。
- 定量脂质体制剂中的未封装药物、辅料和潜在杂质的创新分析方法。
- 鉴别和开发更具预测性、临床相关的体外方法,表征口和鼻用吸入产品雾化颗粒及其沉淀和溶解 ,以及新的计算机建模和模拟方法;作为这项研究的结果,FDA 于 2019 年发布首份基于溶液的计量吸入器(MDI)产品(倍氯米松双丙酸酯) BE 指南。
- 在人类受试者中进行了体内真皮开流微灌注(dOFM)研究,以表征剂量反应关系以及潜在混杂因素,例如相邻治疗位点间探针之间的局部“串扰”,或通过清除作用将药物重新分捕到体循环和皮肤中。
- 开发、评估和改进了基于生理学的药代动力学模型,以应对眼用药、吸入剂和真皮药物仿制药开发给药途径的挑战。
- 定量临床药理学(QCP)方法用于评估稀疏采样 PK 研究设计中的 BE 评价新方法。
- 开展基于机器学习的事件分析,预测涉及新化学实体(NCE)的 ANDA 的首次提交时间。
往期 OGD 年报资讯
作者:识林-椒
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
适用岗位: - “注册”:必须熟悉ANDA的提交流程和要求,以便及时准备和提交增补申请,确保ANDA在最早合法批准日期获得最终批准。
- “QA”:需监控产品质量更新和相关变更,确保ANDA的变更符合FDA要求,并在最终批准请求中明确所有变更。
- “研发”:应关注ANDA的专利声明和专营期影响,以及ANDA的生物等效性更新,确保ANDA的研发符合FDA的最新要求。
工作建议: - “注册”:及时跟踪ANDA的审评状态,合理规划ANDA的增补提交时间,避免因增补导致最终批准延迟。
- “QA”:确保ANDA的质量信息更新及时反映在最终批准请求中,并与原始提交的ANDA进行对比,注明差异及解释。
- “研发”:针对ANDA的生物等效性研究,根据新发布或修订的具体产品指南,及时更新ANDA内容,确保ANDA的科学性和合规性。
适用范围:
本文适用于化学仿制药(ANDA),涉及创新药的专利声明、专营期影响以及ANDA的暂时批准和最终批准流程。适用于美国FDA监管下的化学仿制药企业,包括Biotech、大型药企、跨国药企以及CRO和CDMO等。 要点总结:
本指南旨在指导申请人准备和提交暂时批准ANDA的增补,包括最终批准请求。强调了ANDA批准时间受RLD专利和/或专营期保护的影响,要求ANDA申请人在ANDA中提供与RLD专利相关的信息,并提交相应的专利声明。指南详细描述了ANDA暂时批准后增补的审评目标和状态,以及最终批准请求的提交和审评目标。特别指出,ANDA申请人需在最早合法ANDA批准日期前提交最终批准请求,以确保ANDA能够及时获得最终批准。此外,指南还提供了可能影响最终批准的TA后变更的非完全清单,以及最终批准请求的内容要求,以协助申请人确保ANDA在请求最终批准前是完整且最新的。 以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - 注册(RA):必读。需熟悉ANDA与505(b)(2)申请的区别,以便在药品注册路径选择时提供专业建议。
- 研发(R&D):必读。在药品开发阶段,需明确产品是否符合ANDA要求,或是否需要通过505(b)(2)途径提交。
- 市场(MKT):必读。了解不同申请路径对市场策略的影响,尤其是在专利和独占权方面。
- 临床(Clin):必读。在设计临床研究方案时,需考虑研究结果是否适用于ANDA或505(b)(2)申请。
工作建议: - 注册(RA):在准备药品注册文件时,应评估产品是否符合ANDA的要求,或是否需要通过505(b)(2)途径提交,特别注意药品的生物等效性、活性成分的一致性以及与RLD的差异。
- 研发(R&D):在研发过程中,应密切关注与RLD的差异,确保这些差异不会影响药品的安全性和有效性,并在必要时与FDA沟通。
- 市场(MKT):在制定市场进入策略时,考虑ANDA和505(b)(2)申请对市场独占期的影响,以及如何利用这些信息来优化市场策略。
- 临床(Clin):在设计和执行临床试验时,确保研究设计符合所选申请路径的要求,并在必要时与FDA沟通以确认方案的适用性。
适用范围:
本文适用于化学药品,包括创新药和仿制药,以及生物制品。适用于美国FDA发布的指南,涉及Biotech、大型药企、跨国药企以及CRO和CDMO等各类企业。 要点总结:
本文提供了FDA关于决定提交ANDA还是505(b)(2)申请的指南。ANDA适用于与RLD在活性成分、剂量形式、给药途径、强度、使用条件和标签上相同的药物产品,而505(b)(2)申请则适用于需要依赖FDA对RLD的安全性和/或有效性评估,且至少部分信息来源于非申请人进行的研究。ANDA通常不包含临床研究以证明安全性或有效性,而505(b)(2)申请则可能包含。ANDA必须显示与RLD生物等效,而505(b)(2)申请可能需要额外的数据来支持差异。此外,ANDA的标签必须与RLD相同,除非有专利或独占权的差异。对于可能需要提交超出ANDA范围数据的产品,应考虑505(b)(2)申请,并在提交前与FDA沟通。 以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |