首页
>
资讯
>
EMA发布集中程序获批药品抽样与检验20年报告
出自识林
2019-04-05
3月28号,欧洲药品管理局(EMA)发布了集中审评程序获批药品抽样与检验20年报告[pdf]
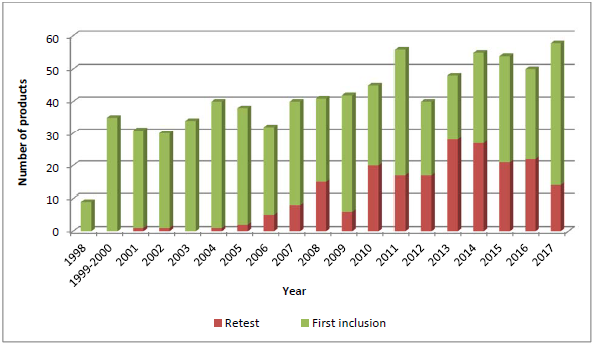
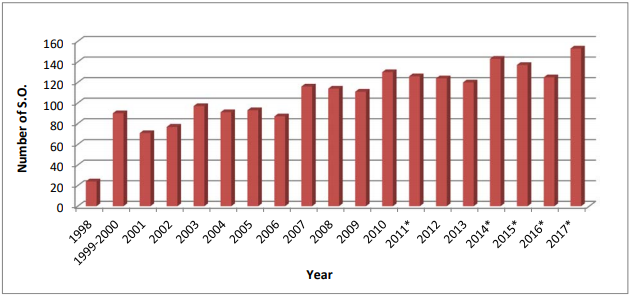
抽样与检验项目旨在监督欧洲市场上的集中审评药品质量,由欧盟委员会(European Commission)、欧洲药品管理局(European Medicines Agency)、欧洲药品质量管理局(European Directorate for the Quality of Medicines,EDQM)和官方药品质量控制实验室网络(network of Official Medicines Control Laboratories,OMCLs)共同发起于1997年,从1997-1998试点计划检验的9个药品稳定增加至2017年的58个,总计检验超过700个药品。EDQM根据检验项目决定取样数量后,从欧盟/欧洲经济区(EU/EEA)三个国家中取样,并交由OMCLs检验,通常化学药品和胰岛素样药品由一个实验室检验,非化学药品由两个实验室检验。
药品选择
1. 基于风险选择药品
自2009年开始,EMA基于风险选择集中审评药品,利用已有信息对产品风险进行排序,筛选排名靠前的品种进行检验 。风险排序需要风险因子识别和对风险因子赋予权重。通常情况下,风险水平与风险因子影响的严重程度有关,也与其发生的可能性有关。EMA成立了由有抽验经验的EMA与OMCL网络中的专家代表组成的咨询组,协助EMA制定抽样计划。
EMA在其他非CAPs抽样计划实施中进行实验,并汲取了一些经验:被评价的风险因子在数量上要少,风险评价 所需的信息应容易获得。风险水平的构成与风险因子影响的严重程度(i)、因子权重值(w)和评价者基于对产品或工艺深入分析基础上赋予的权重值(r)有关,即:风险水平=i*(w+r)。
检验名单由EMA秘书处(EMA Secretariat)和科学委员会(EMA Scientific Committees)准备,再送至EDQM,并由EDQM协调取样和检验。
2008年-2014年的抽验药品名单可依次查看下列文件的附录2(注:EMA官网只公布了2008年-2014年抽样和检验的报告):
Results of the sampling and testing programme for the year 2014 [pdf] Results of the sampling and testing programme for the year 2013 [pdf] Results of the sampling and testing programme for the year 2012 [pdf] Results of the sampling and testing programme for the year 2011 [pdf] Results of the sampling and testing programme for the year 2010 [pdf] Results of the sampling and testing programme for the year 2009 [pdf] Results of the sampling and testing programme for the year 2008 [pdf] 值得注意的是,项目每年检验50-60个药品,包括初次检验和再检验,而再检验通常由于初次检验后生产工艺 发生重大变更 或大量质量相关的上市许可 变更引起。
基于风险选择药品的特殊性在于,在品种上市许可阶段就开始寻找依据,并综合考虑审评官、GMP 检查员的建议进行风险评估。
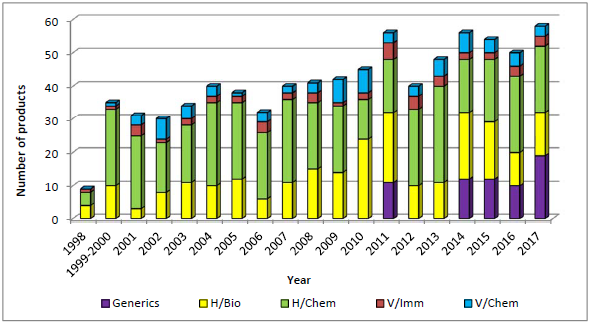
2. 涵盖仿制药、生物制品、化学药品
项目涵盖人用药和兽用药,包括生物制品 和化学药品。值得注意的是,自2014年开始,每年均会对仿制药 进行抽样和检验。
注:H/Bio:人用生物制品;H/Chem:人用化学药品;V/Imm:兽用免疫制品(Imm = Immunological);V/Chem:兽用化学药品
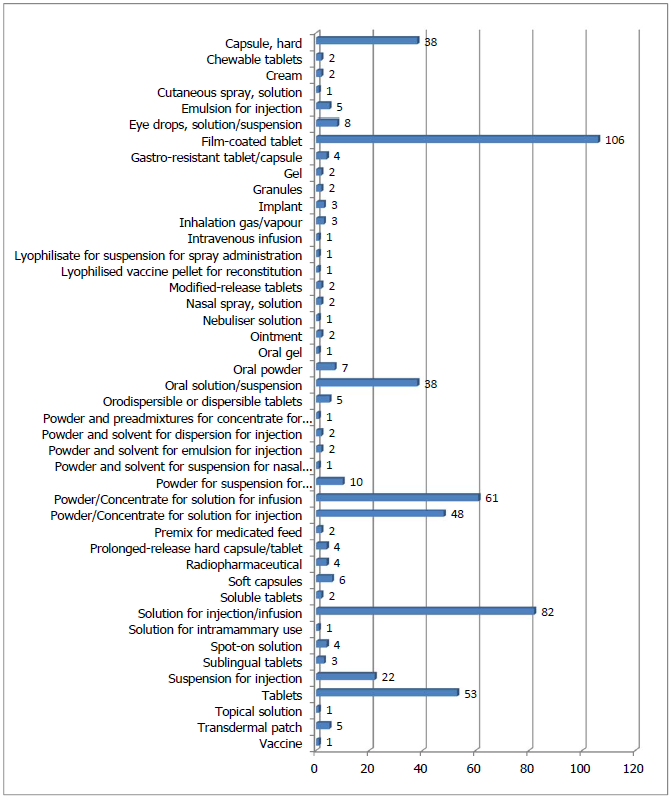
3. 胶囊/片剂、注射用/输液用粉针剂、溶液、悬浮液是最为常见的抽检剂型
图3汇总了各类剂型的抽检数量,其中胶囊 /片剂 、注射 用/输液用粉针剂 、溶液、悬浮液是最为常见的抽检剂型 ,这与每年批准上市的CAP剂型的情况基本一致。
药品取样
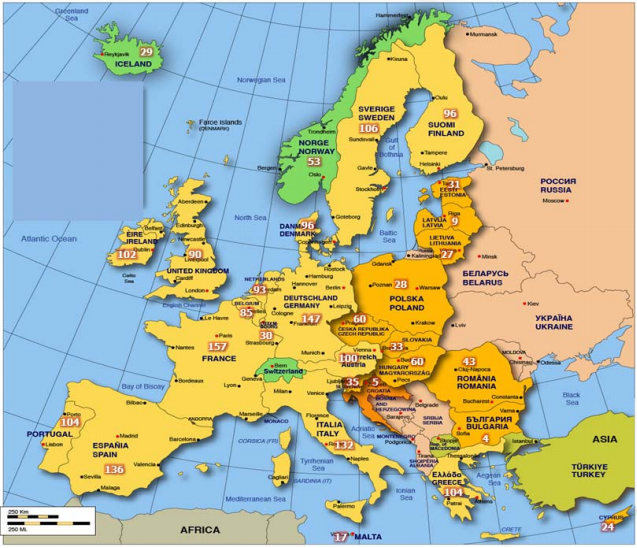
1. 样品来源于EU/EEA市场
项目通常会从三个EU/EEA国家取样,由EDQM决定,考虑的因素包括:各成员国气候条件、共享样品的工作量和药品销售量。
2. 取样量的决定因素众多
取样时,通常从每个国家抽检一个批次的样品(三个国家共三个批次),取样量取决于:每个检验程序要求的药物制剂单元、待检剂型的检验项目量、产品可及性、市场大小、药物的临床应用等。
取样地点由取样成员国决定,通常取决于:产品可得性、EDQM要求的包装量以及取样操作和检查员能力,这也解释了为什么大多数的取样是在经销商或上市许可持有人(MAH)仓库层面上进行。
药品检验
1. 检验项目
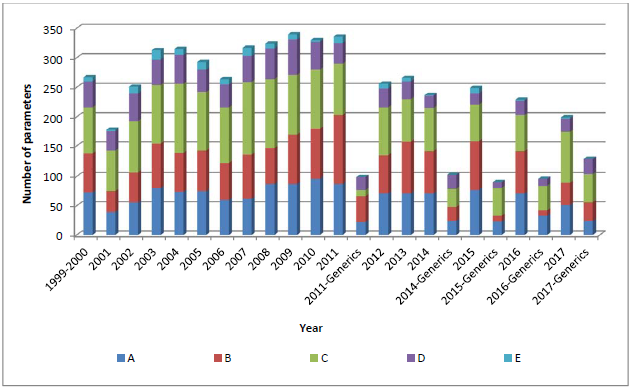
检验项目的选择基于负责审评药品卷宗的专家(Rapporteur and co-Rapporteur)的建议,他们对产品深入的理解使得他们能够选出产品最为关键的检验项目。检验项目通常被分为五类:
Category 分类
Parameters 项目
A
Tests related to the determination of active substance/preservatives content and potency; this also includes the tests for uniformity of content 和原料药 /防腐剂含量 和效价相关的测定,也包括含量均匀度 的检验
B
Tests performed to assess the purity of the medicinal product and/or the integrity of the active substance (e.g. related substances, residual solvents , molecular size distribution) 药物和/或活性物质纯度(如有关物质、残留溶剂 、分子大小分布)的评估
C
All tests linked to physical/pharmaceutical characteristics (uniformity of mass, disintegration of tablets , appearance , colour, clarity ); this category also includes the tests for water content, particulate matters and particle size 物理/制药特性(质量均匀度、片剂崩解 、外观、颜色、澄清度 )相关的检验,也包括水分、颗粒物和粒度 检验
D
Identity tests 鉴别 检验
E
Microbial/bacterial contamination tests such as determination of bacterial endotoxins , sterility test 微生物/细菌污染检验,如细菌内毒素 测定、无菌检验
相比于鉴别(类别D)和微生物/细菌污染检验(类别E),原料药或防腐剂分析(类别A)、药物和/或活性物质纯度(类别B)、物理/制药特性(类别C)是最为常见的检验项目。
2. 检验期
样品检验由不同国家的官方药品质量控制实验室(OMCLs)进行,各成员国实验室进行的检验次数可点击原报告查看。
除样品、标准品 外,OMCLs还会收到MAH的检验方法 ,因此,只需要完成分析方法转移即可,而无需重新进行验证 。化学药品的检验需要在40个工作日内完成,非化学药品则需要在65个工作日内完成。检验时间的延长包括:产品需要大量的检验、检验中包括原料药或检验中遇到了问题等。
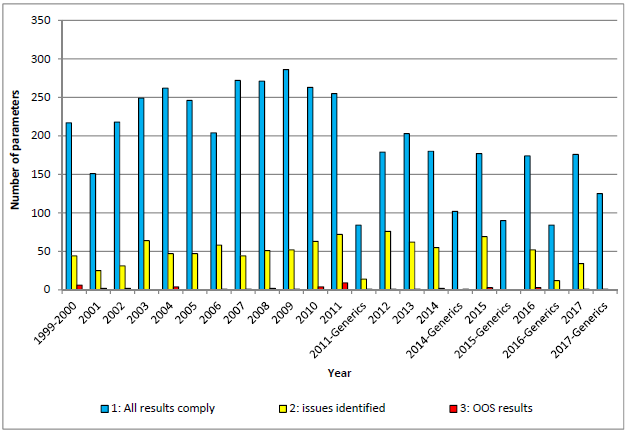
3. 大部分药品检验符合标准
检验结果有以下三种:
所有结果均符合要求-没有发现问题;
发现问题,需要与专家(experts/rapporteur/co-rapporteur)商议;
结果超出质量标准 (OOS );
所有项目的检验结果显示,大部分检验结果符合质量标准。
注:本图为所有项目的检验结果,更多人用/兽用化学药品、人用/兽用生物制品的统计内容请查看原报告。
多数检验中遇到的问题需要MAH、EMA秘书处、审评官、EDQM、检验实验室等多方交流和澄清,某些情况下,MAH将需要变更检验方法或相关SOP。
某些情况下,当超标结果确认后,可能引起其他监管行动,如质量缺陷、重新检验、检查等,至今为止,共4起批次召回发生:
2006年,因生长激素注射液外观 不符合标准;
2009年,因兽用抗体总杂含量不符合标准;
2017年,因唑来膦酸仿制药 批次未知杂质含量超标,以及美洛昔康仿制药pH 值超标;
另外,不同类型产品的检验项目出现技术/科学或监管问题的比例不同:
人用化学药品:9%
兽用化学药品:9%
人用生物制品:18%
兽用免疫制品:25%
这可能与生物制品本身的复杂性和分析方法的复杂性相关。
2019年展望
2017年EMA开始和EDQM以及集中审评药品咨询小组(CAP Advisory Group)共同进一步扩大抽样和检验项目。新的集中审评药品市场监督计划包括:
1. 检验更多生物制品生物类似药 检验项目互认程序 (MRP)/分权审评程序(DCP)的仿制药抽样与检验
作者:识林-栀® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料
[1] 20 years of sampling and testing of centrally authorised products: 1998-2017 [pdf] EMA topic Sampling and testing 20 years of sampling and testing programme for medicines authorised for the EU European Directorate for the Quality of Medicines and HealthCare (EDQM) of the Council of Europe Sampling and testing of centrally authorised products: Development of risk-based approach for the selection of products
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。