首页
>
资讯
>
读17份国家集采“列入违规名单的公告”
出自识林
读17份国家集采“列入违规名单的公告”
2026-06-16
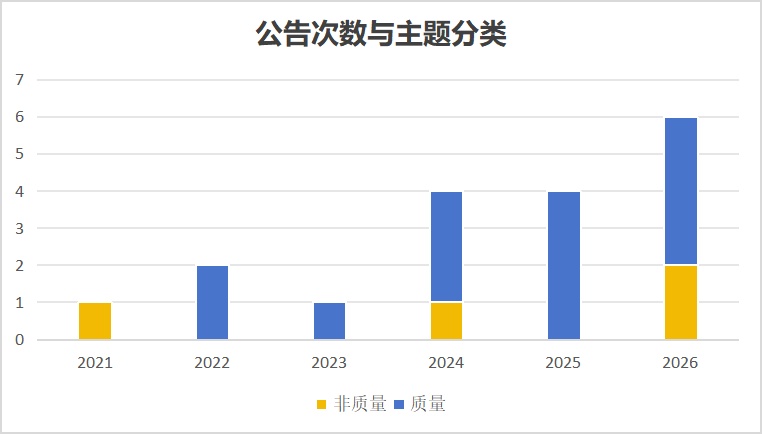
自国家组织药品集中采购(下称“集采”)建立常态化惩戒机制以来,“违规名单”成为评估我国制药行业合规生态与质量管理体系 韧性的实证指引。
截至2026年6月,共计17份“列入违规名单的公告”发布(包含18个违规案例),除了非技术性的供应违约和投标违规,其余14个案例集中于药品生产质量管理规范 (GMP)相关的严重检查缺陷 ,这包括个别外企(韩国DAEWOONG BIO )的拒绝检查,以及宁波大红鹰 案例中“生产劣药”的案例(未找到具体检查缺陷)。
笔者通过识林数据库,检索到对应检查公告中的缺陷描述,形成一份信息汇总表 ,会员可点击查阅并可跳转相关文件。
下文基于表格,对这些案例做简单介绍,供读者参考。如有差错疏漏,感谢指正。
从供应问题到严重质量缺陷
纵观2021年至2026年中上旬的数据分布,违规事件的触发机制经历了从供应保障风险向GMP合规缺陷的递进。
在常态化惩戒机制确立初期(2021—2023年),第一份报告源自2021年华北制药布洛芬缓释胶囊断供事件 ,核心矛盾可能在于企业中选后的商业化产能无法承载协议采购量。随后两年仅1-2起,均与GMP相关,此时缺陷案例涵盖硬件维护 、清洁 、质量控制 、人员 配置及无菌 保障等方面。
2024至2026年,违规事件的发生频率上升。仅2026年前6个月,因GMP不合规而被列入名单的事件已达4起(涉及重庆德润笙 、广州合和 、哈尔滨力强 ,以及最新的山东北大高科华泰 )。此时的GMP 缺陷案例多为质量体系运行与数据可靠性 缺陷,涵盖偏差调查 、放行 管理、电子数据管理、受托方 监控及记录审核等多个方面。基于动态检查与有因检查的合规缺陷已成为触发集采惩戒的主导因素。
质量案例中MAH和设施所在地
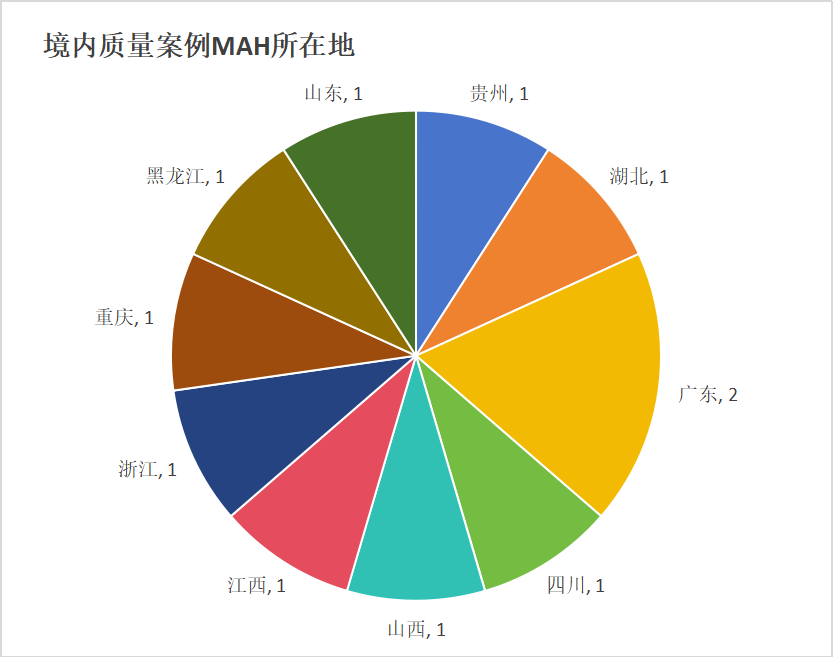
质量相关的14个案例中,剔除境外企业后,余下的11起国内案例中MAH的地理分布整体呈现出较为平均的分散状态。在所有涉及的省市中,仅广东省作为MAH归属地出现了2次(珠海和凡 、广州合和 ),其余包括黑龙江、浙江、江西、山西、湖北、四川、重庆、贵州、山东在内的9个省市均仅各出现1次,未表现出明显的地域性集中趋势。
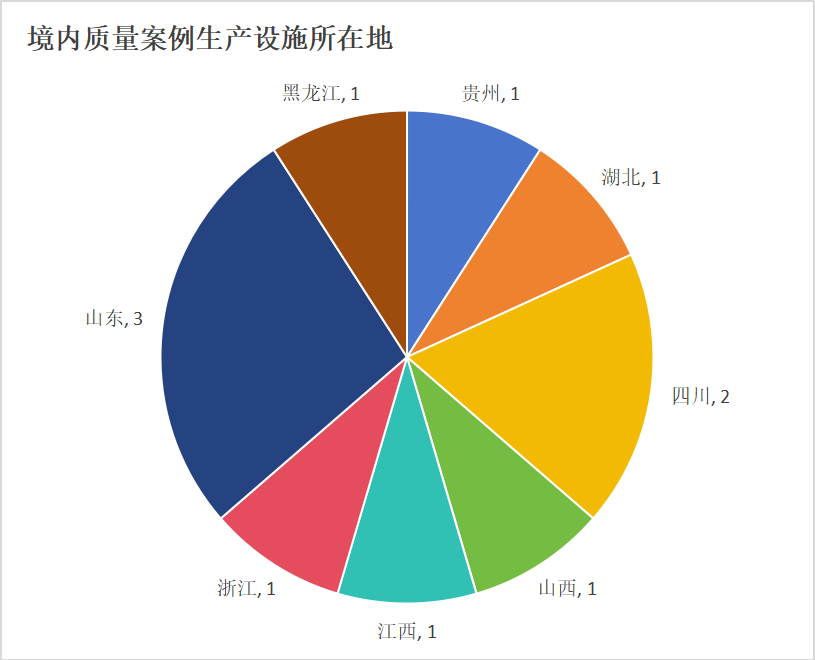
生产设施 所在地药监相应承担生产现场检查职责。其中山东局出现3次(山东威高,潍坊中狮,山东北大高科华泰),四川局2次(四川太极,成都天台山);湖北、山西、江西、浙江、黑龙江、贵州省局则各承检1次。作为MAH归属地出现的广东2次以及重庆的1次记录,均属于委托生产 模式,其品种的现场检查均发生在外省设施(由山东局和四川局承检);值得注意的是,外省生产设施检查后,MAH 所属的广东、重庆两局也开展了针对性检查,并公告不合规结果,但未提具体缺陷。
从口固到无菌,再到复杂制剂
在所有因生产缺陷导致不合规的案例中,受惩戒品种的剂型 分布呈现出明显的侧重。
结合违规公告和检查公告看,注射剂 (包含小容量注射液 、大容量注射液 及无菌粉针剂 )在质量相关案例中占比 64.2%(9/14)。这种高度集中的剂型分布体现出监管部门对无菌品种的特别重视。
可以看到,监管视野在2026年已进一步延伸至吸入制剂 (重庆德润笙的富马酸福莫特罗吸入溶液 )与外用特殊制剂(哈尔滨力强的洛索洛芬钠凝胶贴膏 ),体现出监管部门对集采品种的质量核查已实现全制剂品类的覆盖,囊括冷门或高技术壁垒剂型。
6个案例涉及委托生产
特别值得注意的是,质量案例中共计 6 起涉及委托生产的违规案例,且集中在近两年:
这些案例凸显了MAH制度在落地中的结构性风险。在轻资产运营模式下,部分中选持有人将自身定位为纯粹的商业实体,在通过委托生产契约转移制造风险的同时,可能弱化自身作为质量第一责任人的责任意识。事实上,上述6个案例的缺陷项都在C证企业被检查时发现,但B证企业仍需负主体责任。
从缺陷项描述来看,四川海梦智森 案例中“未能对受托生产企业生产过程进行有效监控”,以及山西阳和 案例中“关键生产工序记录不完整,未对生产过程记录进行有效审核”,均是持有人主体责任虚化的典型表征。持有人对受托方的审计流于形式、关键工艺偏差知情滞后、批记录 审查脱节,导致持有人无法真正掌握药品的质量全貌。这种合规管理的断层在集采连续生产的压力下可能演变为系统性缺陷。
缺陷项关键词是数据可靠性和偏差调查
尽管官方发布的检查缺陷较为简单,但通过对 14 起 GMP 不合规案例的缺陷项原文进行聚类与分析,还是有一定指导意义。
可以看到,除极个别案例表现为现场管理缺陷——如贵州圣济堂 在“厂房维护、设备清洁、稳定性考察等方面存在严重缺陷”,以及湖北科益 在“质量管理人员配备、无菌保障等方面存在严重缺陷”外,其余核心技术不符合项聚焦于数据可靠性 失效与偏差调查 不充分。
数据可靠性
自2024年末至2026年中,数据可靠性缺陷反复出现,且多与电子化数据相关。
珠海和凡 案例的缺陷描述明确指出“部分电子数据未能采用可靠方式进行记录,不利于追溯产品的生产历史和质量相关情况”。
重庆德润笙 案例与广州合和 案例的缺陷特征类似,分别是“吸入溶液制剂无菌质量管理及无菌保障措施不严格,个别设备电子数据管理存在问题”以及“个别检测仪器电子数据管理存在问题”,均被判定为严重缺陷。
此外,山西阳和 案例中“帕拉米韦注射液关键生产工序记录不完整,未对生产过程记录进行有效审核”这一缺陷,亦属于数据可靠性范畴。
偏差调查
在集采品种追求成本效益与高强度运转的背景下,生产线的周转速率不断压缩,可能导致质量体系中偏差与CAPA (纠正与预防措施)机制流于表面。
四川海梦智森 案例的缺陷原文暴露出持有人与受托方的协同盲区:“持有人未能对受托生产企业生产过程进行有效监控,受托生产企业部分批次产品关键生产过程出现偏差,未按规范要求开展偏差处理”。
2026年6月最新通报的山东北大高科华泰 案例缺陷是:“企业对生产过程中发生的偏差调查评估不充分,偏差发生后未全面评估质量风险,风险管控及处置措施不够完善”。
质量标准与放行管理
除了上述两个关键词外,其余缺陷项还映射了企业在物料全生命周期 与最终放行关口上的管控失效:
综上,集采“违规名单”日趋频繁的更新,可以视为是国家药监局与各省、直辖市药监局对集采药品质量强监管意志的集中体现。国家局与各省局的监督检查、飞行检查 以及有因检查,均旨在确保控费降价后的药品质量安全不打折扣。其中特别是对于采取“B证+C证”委托生产 模式的集采药企,监管机构的态度鲜明:持有人是药品质量的全生命周期第一责任人。持有人和受托方双方的质量管理体系 必须深度融合,共同捍卫质量红线。
作者:识林-实木
识林® 版权所有,未经许可不得转载。
必读岗位及工作建议:
QA(质量保证):必须了解并确保生产过程符合《药品生产质量管理规范(2010年修订)》的要求。 注册:需关注国家药品集中采购政策变化,及时更新申报材料,确保合规。 生产:应按照药监局要求,对产品采取暂停生产、暂停销售的措施,并进行整改。 适用范围:
文件要点总结:
生产管理违规: 明确指出珠海和凡医药股份有限公司吡拉西坦注射液在生产管理方面存在严重缺陷,违反了《药品生产质量管理规范(2010年修订)》。中选资格取消: 由于违背申报材料中的承诺和违反相关条款,珠海和凡医药股份有限公司吡拉西坦注射液的中选资格被取消。违规名单列入: 企业被列入违规名单,暂停其自2024年11月29日至2026年5月28日参与国家组织药品集中采购活动的申报资格。替补程序启动: 对于吡拉西坦注射液主供省份,需启动替补程序,由备供企业替补成为主供企业,并按中选价格供应。供应需求满足: 当备供企业不能满足供应需求时,由所在省份启动增补备供企业供应流程。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证):负责确保企业生产过程符合GMP要求,监督整改措施的实施。 生产(Production):需要了解检查结果,确保生产过程符合规定。 注册(Regulatory Affairs):关注检查结果对药品注册的影响,准备相关文件和报告。 研发(R&D):对于涉及特殊药品、放射性药品和集采品种的企业,需关注检查结果对产品研发的影响。 工作建议:
QA:根据检查结果,更新质量管理体系,确保持续合规。 生产:对于不符合要求的情况,立即采取措施整改,并报告整改进度。 注册:对于检查中发现的问题,准备相应的补充申请或变更申请。 研发:针对检查中提到的特殊药品和集采品种,调整研发计划,确保符合监管要求。 文件适用范围:
文件要点总结:
监督检查结果 :12家药品生产企业接受了监督检查,大部分企业符合要求。特殊药品和放射性药品 :涉及特殊药品和放射性药品的企业需特别注意合规性。集采品种 :部分企业涉及集采品种,需确保生产过程和产品质量符合高标准。不符合要求情况 :山东威高药业股份有限公司因电子数据记录问题被判定为不符合要求,需整改。风险防控措施 :对于不符合要求的企业,采取了约谈、暂停生产等风险防控措施。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。 适用范围:
文件要点总结:
质量管理体系概述 :明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。产品质量实现要素 :涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。质量保证要素 :包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。质量风险管理 :介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。质量管理系统文件 :规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。