|
首页
>
资讯
>
近期三分之二罕见病药物仅依赖一项临床试验加确证性证据获FDA批准
出自识林
近期三分之二罕见病药物仅依赖一项临床试验加确证性证据获FDA批准
2025-03-26
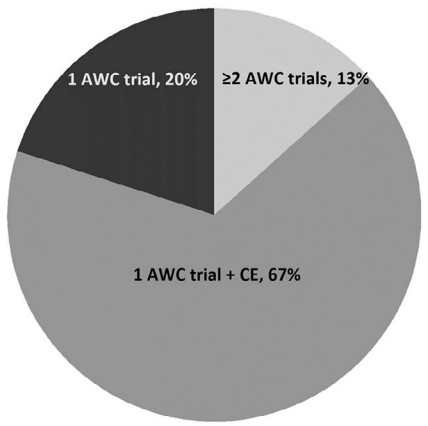
FDA人员对2020-2023年批准的新分子实体(NME)非肿瘤罕见病上市申请的分析发现,在确定的60个获得孤儿药认定的NDA和BLA申请中,40个申请(67%)利用1项充分且对照良好(AWC)的临床试验加上确证性证据来满足FDA的有效性实质性证据(SEE)的要求;12个申请(20%)利用1项极具说服力的AWC试验满足SEE要求;只有8个申请(13%)提供了2项或更多AWC试验。同期获批的用于治疗常见疾病的非肿瘤NME药物申请,71%(49/69)提供了2项或更多AWC试验。
有效性实质性证据(SEE)是NME NDA和BLA上市申请的必要条件,其重要特征包括科学证据的质量和数量,建立SEE通常需要2个或更多AWC临床试验的数据来证明。在某些情况下,1项AWC试验和确证性证据 (CE) 足以建立SEE。有时,单个大型多中心AWC试验在科学和法律上都可以被视为是实际上的多个试验,并且可以依靠它来证明SEE。关于实质性证据和确证性证据的要求,请参见识林主题词【确证性临床试验】和【实质性证据】。
*表格截取自识林主题词【实质性证据】
在2023年9月,FDA发布了 《通过一项设计充分且良好对照的临床研究和确证性证据证明有效性的实质性证据》(Demonstrating Substantial Evidence of Effectiveness Based on One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence),描述了上市申请中常用的确证性证据的类型:
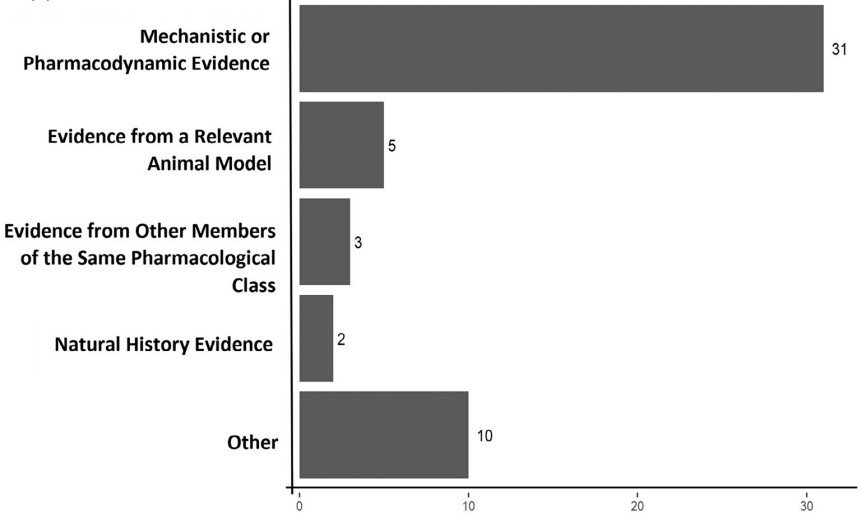
根据指南中描述的CE类别,将上述40个上市申请中所使用的CE进行分类。10项(25%)申请使用了多种CE类型;31项(77.5%)申请使用的是机制或药效学证据,为最常见的类型;相关动物模型的证据(5项,12.5%),来自同一药理学类别其它成员的证据(3,7.5%),自然史证据(2,5%)。
机制和药效学证据通常包括药效学生物标志物和作用机制数据,尽管药代动力学/暴露-反应数据和体外研究信息也偶尔被引用。本分析中的10个上市申请中包含的确证性证据与指南草案中描述的类别不符,最常见的形式是在关键有效性试验之外收集的额外临床证据。
用于支持上市申请的CE类型应根据每个药物开发项目的具体情况逐案确定,并应与相关FDA审评部门讨论,最好从pre-IND阶段开始,直至申请提交。正如FDA指南草案中所讨论的,一项强有力且有说服力的AWC临床研究可能需要较少的CE,而说服力较弱或规模较小的试验可能需要更多有说服力的CE以满足FDA对SEE的要求。
此外,在同样的数据面前,不同专家对于这些数据成为何种证据的理解也有所不同。在2023年12月获批的Filsuvez(桦木三萜)凝胶案例(NME,孤儿药认定)中,FDA审评小组、负责处理上诉请求的副主任和申请人三方,对此项NDA申请中涉及到的4项临床试验提供的实质性证据和确证性证据的理解都有所不同。
Filsuvez在首轮审评中被签发完全回应函,指出该申请在有效性方面的证据不足,为了解决这个问题,需提交额外的确证性证据。申请人通过正式争议解决请求 (Formal Dispute Resolution Request) 提出上诉,上述结论也在正式争议解决流程中被推翻,进而得以获批。案例详情请参见识林案例《通过正式争议解决请求程序,推翻完全回应函中的结论》。助力药企加速创新药上市,保障患者用药可及性。
识林-樟
识林®版权所有,未经许可不得转载
适用岗位: - 注册(RA):必读。需理解指南内容,以便在药品注册过程中准确应用,特别是在准备和提交新药申请时。
- 临床(Clin):必读。在设计和执行临床试验时,需参考本指南以确保试验设计和结果能够支持药品的有效性证明。
- 研发(R&D):必读。在药物开发计划中,需考虑本指南的建议,尤其是在考虑使用单一充分和良好对照的临床研究和确证性证据来证明药品效果时。
工作建议: - RA:在准备注册文件时,特别关注如何通过单一充分和良好对照的临床研究结合确证性证据来满足FDA对药品效果的实质性证据要求。
- Clin:在设计临床试验时,考虑如何通过试验设计和终点选择来增强单一临床研究的说服力,以及如何选择合适的确证性证据。
- R&D:在药物开发早期,与FDA沟通,讨论计划中的单一临床研究和确证性证据,确保开发计划能够满足FDA的要求。
适用范围:
本文适用于化学药和生物制品,包括创新药、仿制药、生物类似药及原料药,由美国FDA发布,适用于Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。 要点总结:
本指南提供了关于如何通过单一充分和良好对照的临床研究结合确证性证据来证明药品效果的FDA当前思考。它强调了在评估药品效果时,单一临床研究的设计特征和结果对所需确证性证据的数量和质量有显著影响。确证性证据可以来自多种来源,包括相关适应症的临床证据、机制或药效学证据、相关动物模型的证据、同一药理类其他成员的证据、自然病史证据、真实世界数据/证据以及扩大使用研究药物的证据。本指南强调了与FDA早期沟通的重要性,以便评估单一临床研究和确证性证据是否足以证明药品效果。此外,指南指出,尽管单一临床研究和确证性证据可能证明效果,但可能需要额外的安全性数据来支持药品的安全性评估和风险效益分析。 以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - 注册部门:深入了解FDA关于证明药品和生物制品有效性的指导草案,以便在新药申请(NDAs)和生物制品许可申请(BLAs)中准确呈现有效性证据。
- 研发部门:掌握临床试验设计、终点选择和统计考量,确保临床研究的质量和结果的科学性。
- 临床部门:熟悉临床证据的质量要求,包括试验设计、终点和统计分析,以指导临床试验的规划和执行。
文件适用范围:
本文适用于化学药和生物制品,包括创新药和仿制药,由美国食品药品监督管理局(FDA)发布,主要针对大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业。 文件要点总结: 实质性证据标准:强调药品和生物制品必须通过“实质性证据”来证明其有效性,这通常需要充分和受控的临床研究。 临床证据的质量:讨论了试验设计、试验终点和统计考量对临床证据质量的影响,以及如何通过这些因素来确定有效性。 临床证据的数量:详细说明了基于两次充分和受控的临床研究、一次加上确认性证据或依赖于FDA之前对已批准药品有效性发现的证据来满足实质性证据标准的情形。 特殊情况下的灵活性:在疾病危及生命或严重致残且医疗需求未得到满足、疾病罕见或进行人体有效性试验不道德或不可行的情况下,可能需要额外的灵活性。 安全性与有效性的综合考量:虽然实质性证据的发现对FDA批准至关重要,但批准决定还需要确定药品对预期使用安全,这涉及到对药品风险和益处的综合评估。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - 注册(RA):必读。需理解文件要求,确保申报资料符合FDA对临床证据的要求。
- 临床(Clin):必读。在设计临床试验和解释数据时需遵循指南原则。
- 研发(R&D):必读。在药物开发阶段考虑临床证据的有效性要求,规划研究策略。
工作建议: - 注册(RA):在准备NDA或BLA时,参照本指南确保所提交的临床数据满足FDA对有效性证据的要求,并在申报策略中体现。
- 临床(Clin):设计临床试验时,考虑如何通过一项或多项研究提供充分的有效性证据,并注意多中心研究的内部一致性。
- 研发(R&D):在早期研发阶段,考虑如何通过不同研究设计和终点来支持新用途的有效性,以及如何利用已有数据支持新申请。
适用范围:
本文适用于化学药和生物制品,包括创新药、仿制药及生物类似药等。适用于在美国进行注册的大型药企、Biotech公司以及跨国药企。 要点总结:
本指南明确了支持药品和生物制品有效性所需的临床证据数量和质量标准。法律标准要求通过充分和良好控制的研究来证明药品的有效性,通常意味着至少需要两项令人信服的研究。然而,FDA也灵活处理,允许在特定情况下依靠单一研究的证据。这些情况包括:从现有研究中推断新用途的有效性、单一研究的新用途得到相关研究数据的独立支持、以及单一多中心研究在没有其他研究支持的情况下提供有效性证据。指南强调了独立证据的重要性,以避免单一研究中的偏差、偶然性或欺诈性结果导致错误的有效性结论。同时,指南也讨论了在提交补充申请时,如何利用已提交的数据支持新的适应症。此外,还涉及了在数据质量证明方面,对于不同类型的研究和数据,FDA可能接受不同程度的文件记录。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |